Introduction
Input
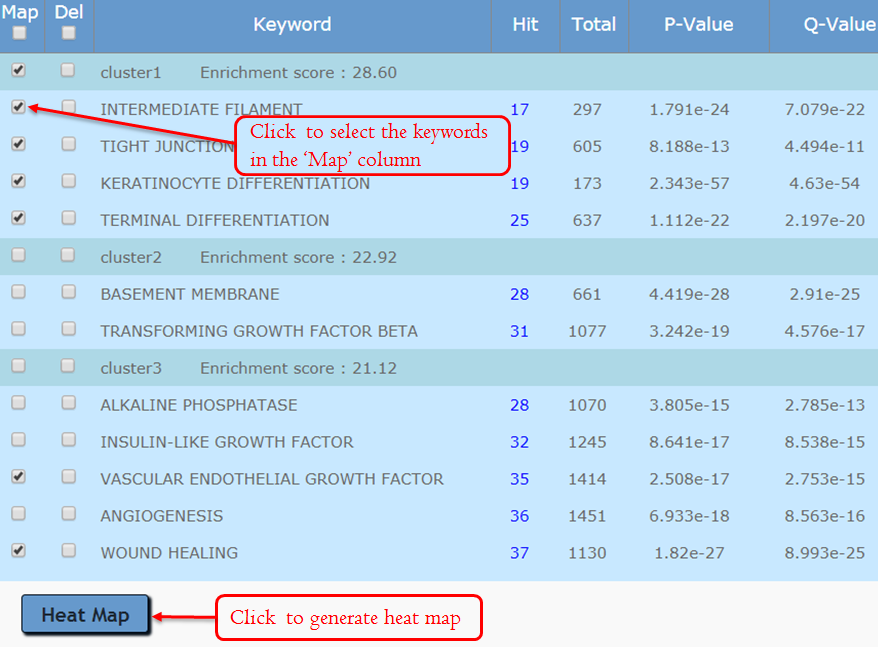
Function Enrichment with Literature Keywords
Gene Network Analysis
Gene Network Search
Function Related Gene and Gene Network
GO / Pathway Analysis
Fuzzy Cluster
Login/Register
Introduction
We updated our software GenCLiP to GenCLiP 3. As compared with previous versions and the other similar text-mining tools, the unique characters of GenCLiP 3 are: (i) integration of CoreNLP to accurately recognize molecular interactions and their interaction polarity and directionality with Semgrex patterns from the entire PubMed database; (ii) integration of Sphinx with MySQL to support Boolean search and to quickly retrieve function-related genes from more literature sources; (iii) identification of gene-related keywords by a new scoring method that considers the co-occurrence of a gene and keyword in a sentence and abstract; and (iv) daily updates following the release cycle of PubMed FTP.The functions of GenCLiP 3 include: (i) annotation of a gene list with enriched keywords (pre-defined or provided by a user) from gene-related literature, or with enriched GO terms and pathways; (ii) creation of a comprehensive gene regulatory network for a gene list with text-mined and curated interactions; (iii) search direction and indirection interactions between two genes; and (iv) a retrieve function for related genes by querying a co-occurring gene with complex and custom search terms, and creation of a gene regulatory network for related genes.
For analysis, the user needs to upload a list of gene IDs or terms. Function Enrichment with Literature Keywords module will generate statistically over-represented keywords grouped by a fuzzy cluster algorithm to annotate the input genes. Users can manually edit these keywords. Gene Network Analysis and Gene Network Search modules will construct gene regulatory networks for a gene list and two genes, respectively, based on literature mining and/or curated databases. Cytoscape.js has been integrated to enhance the performance of interactivity and visualization. Function Related Gene and Gene Network module can query genes co-occurred with terms in the literature with or without several limitations (such as impact factor). The user can build the search using Boolean operators to combine any terms to retrieve function-related genes efficiently and effectively. Besides, GenCLiP 3 integrated GO and pathway databases to annotate the gene list.
Input
GenCLiP 3 accepts four types of gene accessions/IDs. Users can view all the gene accession options from the drop-down selection menu at the input page.
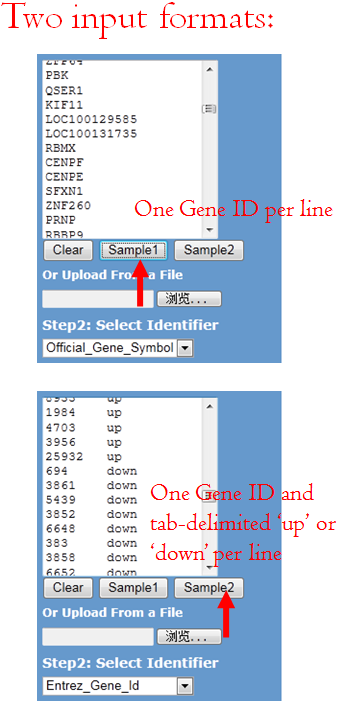
You can either load a gene list from a .txt file or paste a gene list to the text box. Two formats are supported:
1. One Gene ID per line, refer to Sample 1.
2. One Gene ID and tab-delimited ‘up’ or ‘down’ per line, refer to Sample 2.
For user-defined up- and down-regulated genes, the software distinguishes gene name in color, red represented up-regulated, green represents the down-regulated gene.
Please note that, a gene list that over 3000 genes is forbidden.
For registered users, name your job if you wish, that name can help you to retrieve the job later easily, or the system will automatically name it by a random number and the time you submitted. Unregistered users have no permission to retrieve former analysis, so ignore this option.
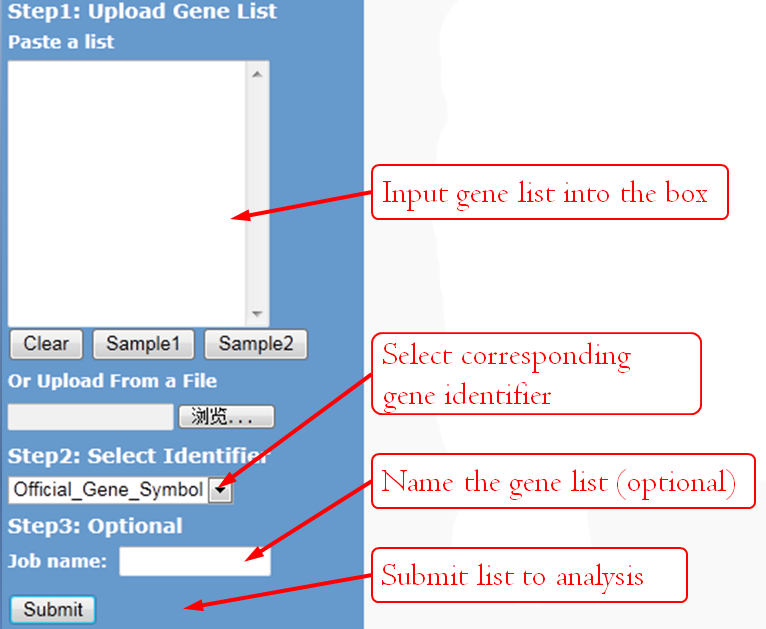
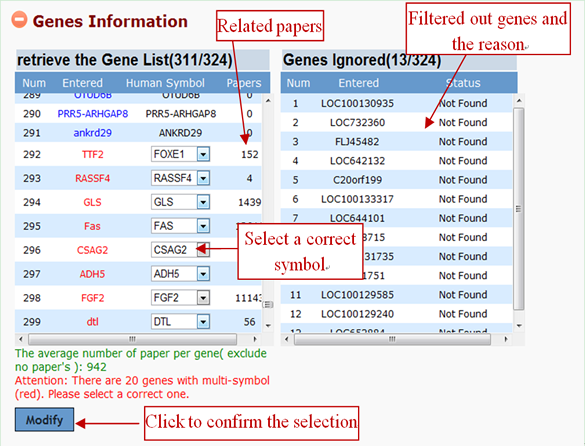
After the gene list is submitted to GenCLiP 3, the system will screen the database and return the gene information. It is able to attach meaning to a list of gene accessions/IDs by rapidly translating them into their corresponding symbol, and to sort input genes alphabetically in the information box. Please note that many gene synonyms are ambiguous, thus one and the same synonym is often used for different genes. Users can select a correct gene through the drop-down selection menu, and click the “Modify” button to change.
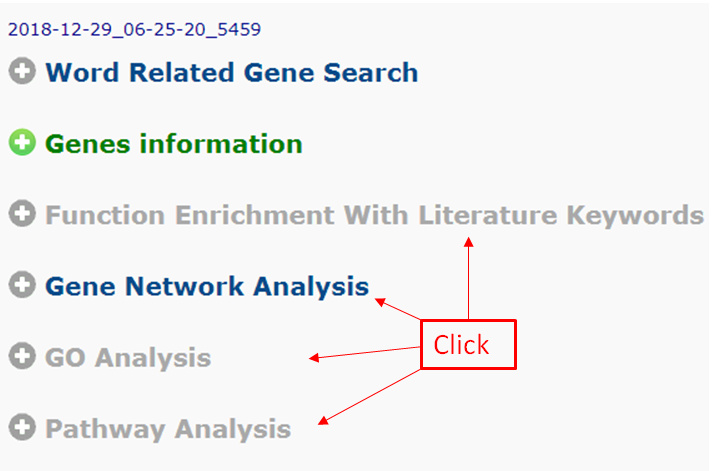
Click the the module to begin further analysis.
Function Enrichment with Literature Keywords
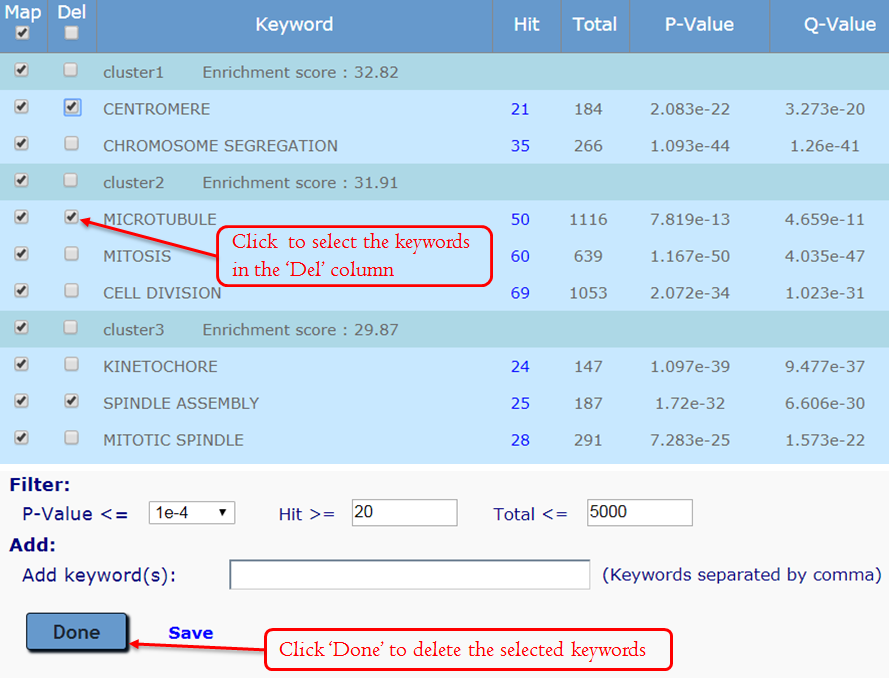
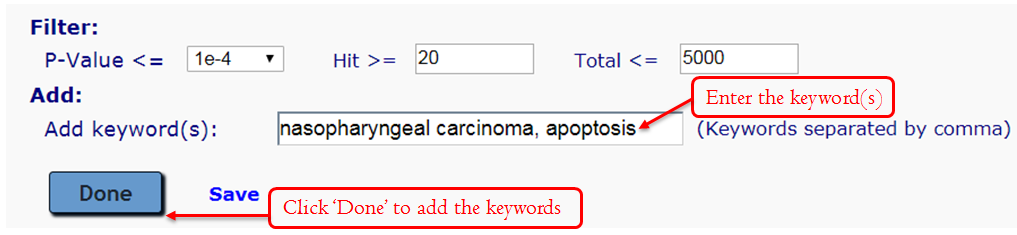
This module focuses on gene annotation based on the free word. There are two ways to apply keywords, automatic and manual. The user can add relevant keywords and remove inappropriate keywords. The purpose of keywords’ editing is to allow you to find the topics that relate to a particular part of your research.When the user clicks the grey icon, the system will extract the keywords automatically. After several seconds’ processing, this section will be expanded, and the annotation table and the optional panel will be shown. For unexpected keywords, users can remove them. For expected keywords but cannot be generated automatically, users can add them manually. The keywords’ enrichment results are classified to multiple groups clearly and concisely using a fuzzy cluster algorithm (see the detail at Fuzzy Cluster ).
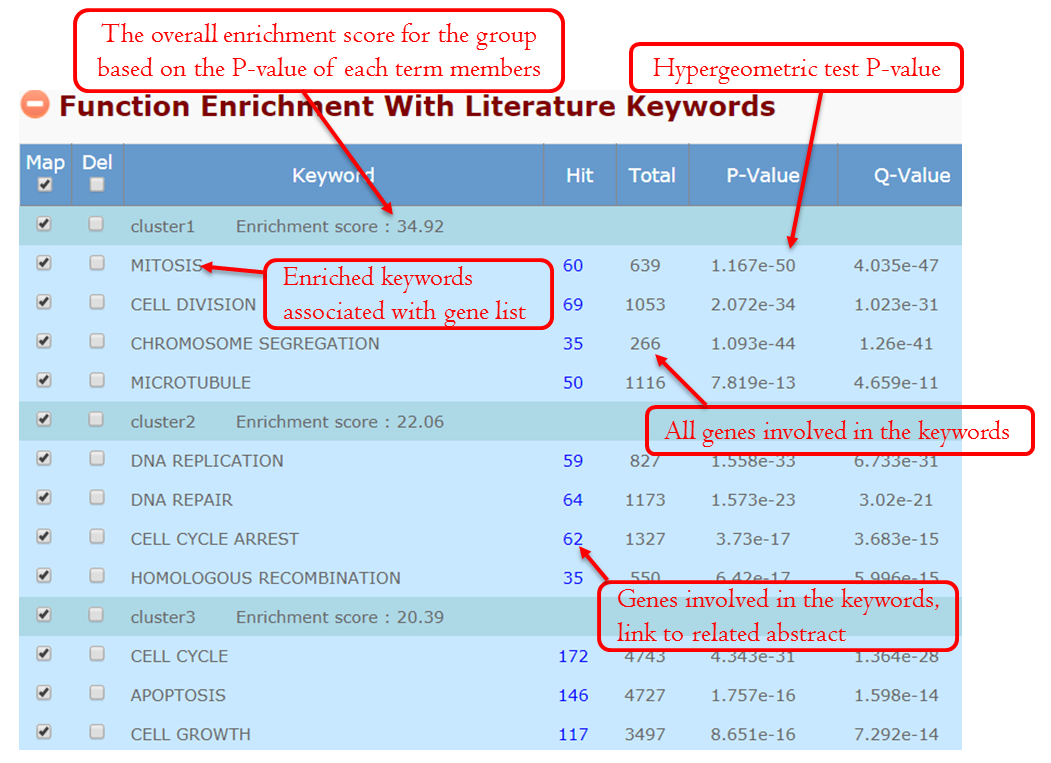
Manually editing – Filter Keywords:
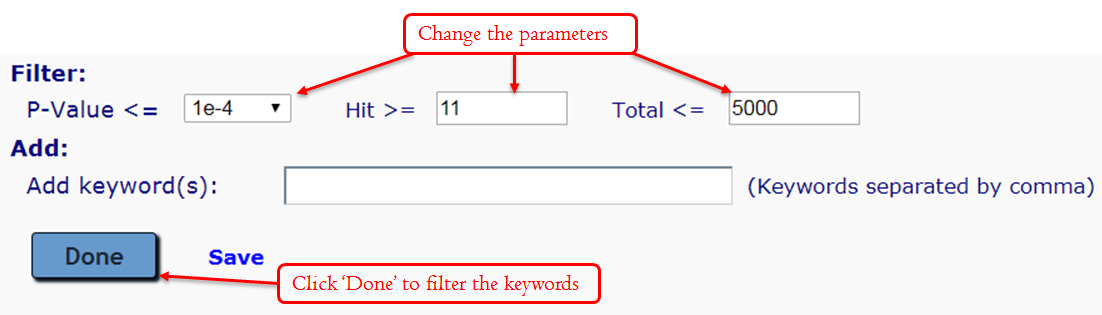
Manually editing – Delete Keywords:
Manually editing – Add Keyword:
To save your annotated result from your browser to your hard drive simply click the link “Save” then type your filename and save to your hard drive. You can then open this file in Microsoft Excel.
Cluster Analysis
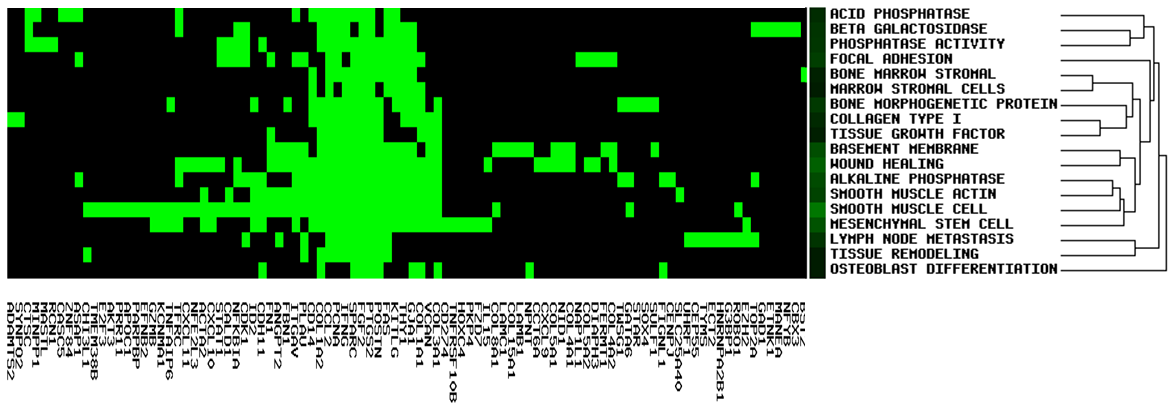
GenCLiP 3 uses the gene list and selected annotations, including keywords, Go terms and pathways, for clustering analysis using the average linkage hierarchical clustering algorithm. The cluster results will be displayed in the image box and can be checked easily. Users can download the result txt file and input for public software, such as Cluster/Treeview programs.
It allows users to see gene members and their associated annotation term in a heat map type of view so that the user can further explore the gene-gene and term-term relationships. This function is also provided for examining the internal relationships among the terms and genes.
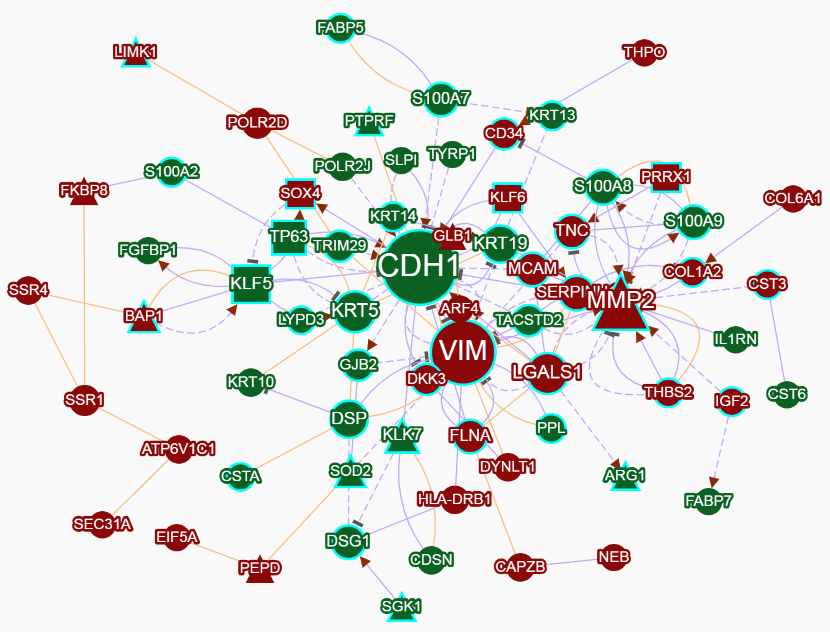
Gene Network Analysis
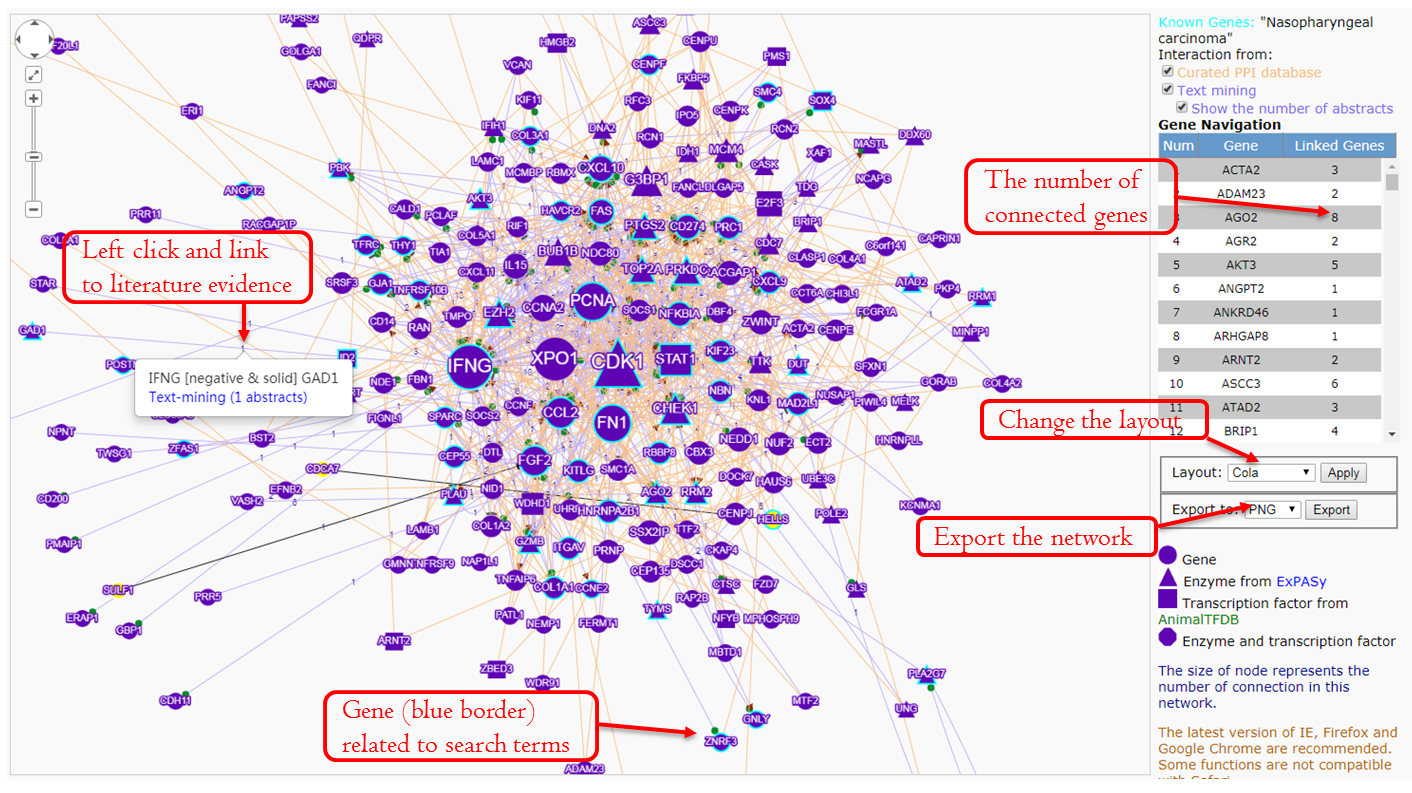
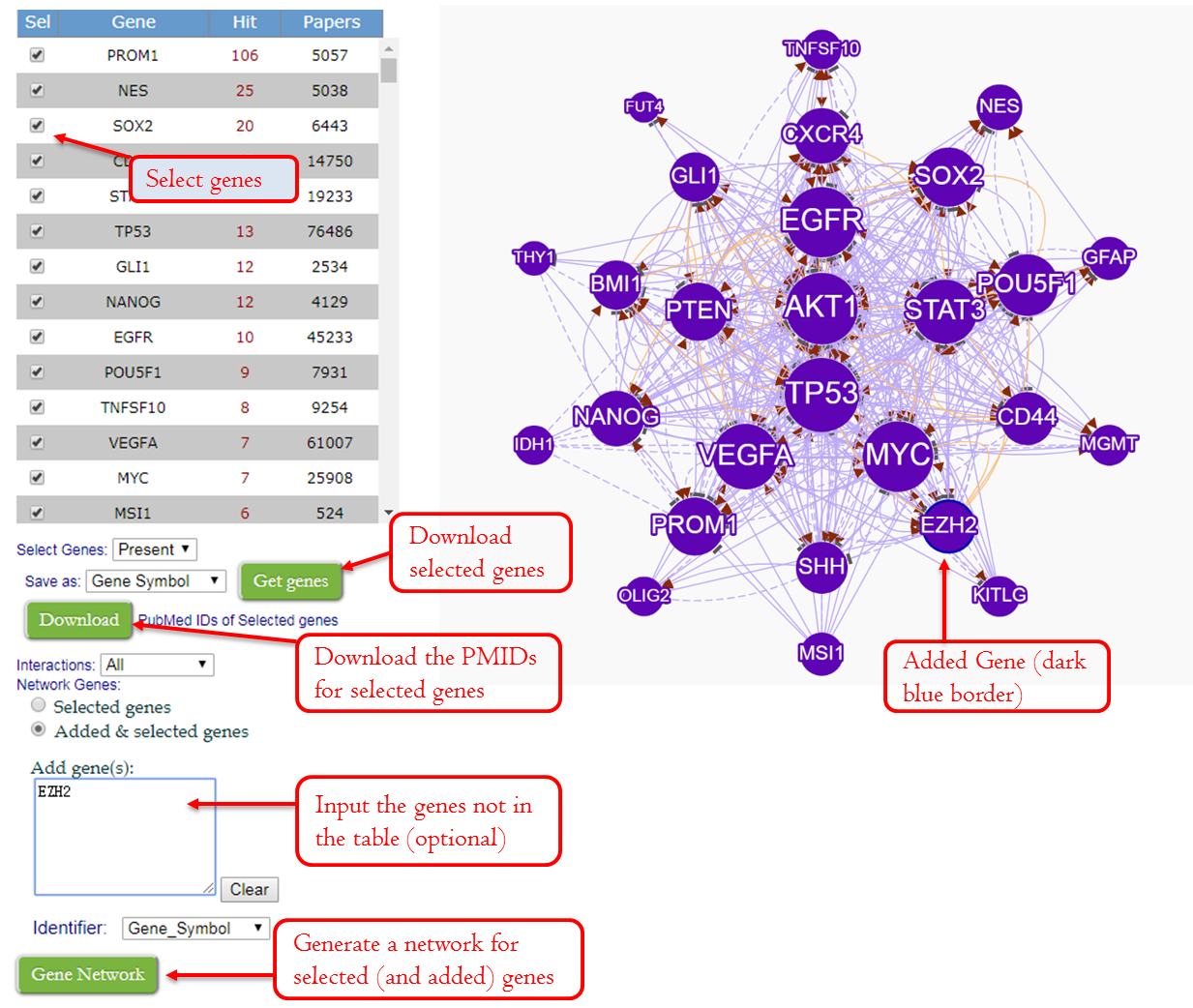
This module can generate a gene network with interaction pairs that are recognized by the Semgrex pattern (Text mining) or collected from the public PPI database (Curated PPI). The search terms in the input box are used to decide whether one gene in the network will be highlighted in a blue border (related to the input terms) or not. Here, Boolean search is supported. Genes are considered to relate to the terms if they co-occurrence in a sentence.

In the network, Different nodes represent different gene functions, Different edges represent the interaction polarity and directionality.
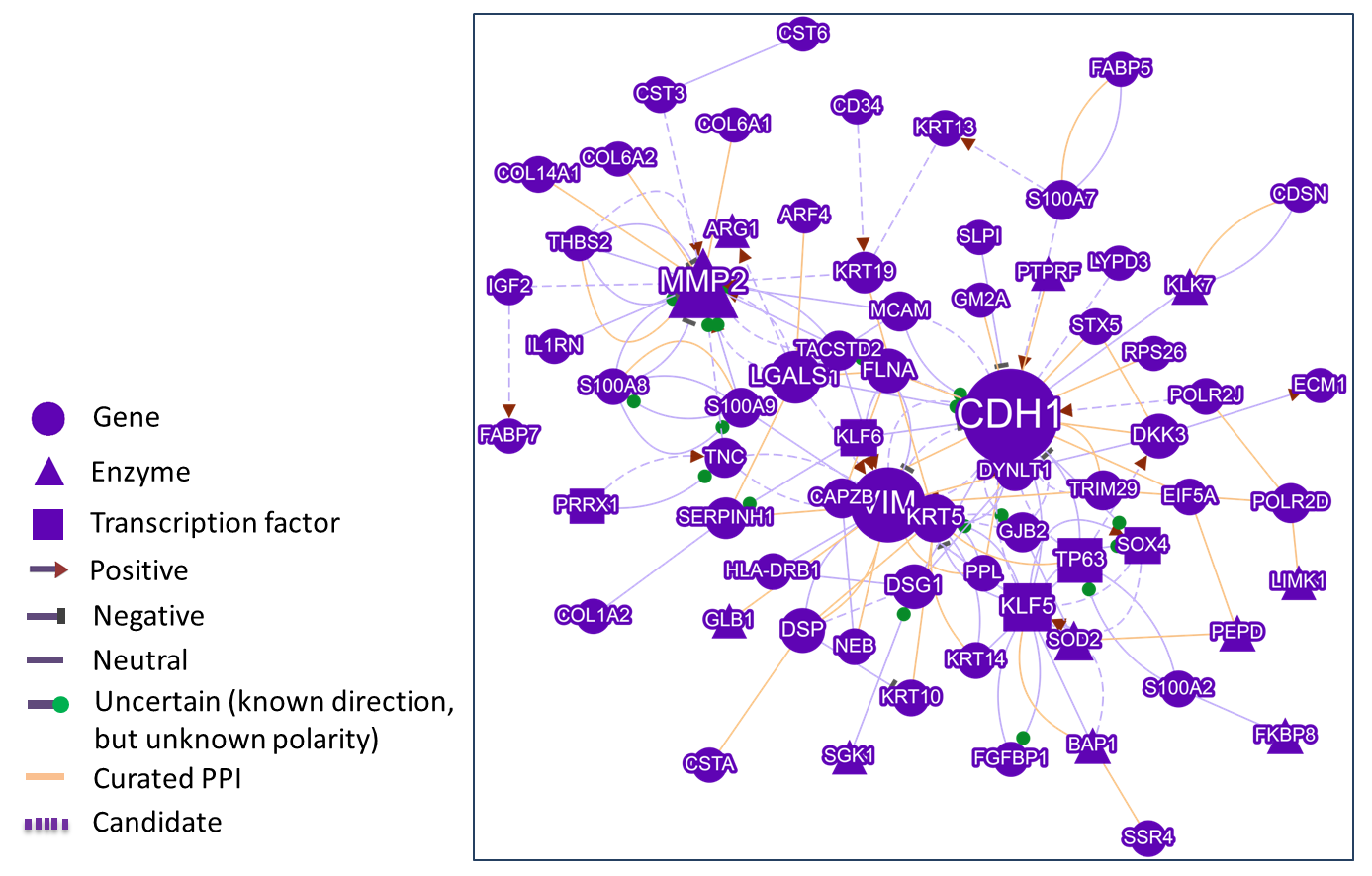
The network is drawn by Cytoscape.js, with Context menu and Panzoom extensions.
- Use the Layout menu to view different arrangements of the network. We set Cola Layout as default.
- Select the file format, click the Export button to save the file to the local disk.
- Right-click the edge or number and click ‘Interaction Detail‘ can link to the literature evidence.
- To simplify the network or remove the wrong gene pair, users can delete the genes and the edges. Right-click a gene and Remove Node can remove this gene and related connections. Right-click an edge and Remove Edge can remove this connection.
For up- and down-regulated genes.
Gene Network Search
The two genes may interact directly or interact through a mediator. This module is designed to help the users to find the direct or indirect interactions of two input genes, and to create a gene network.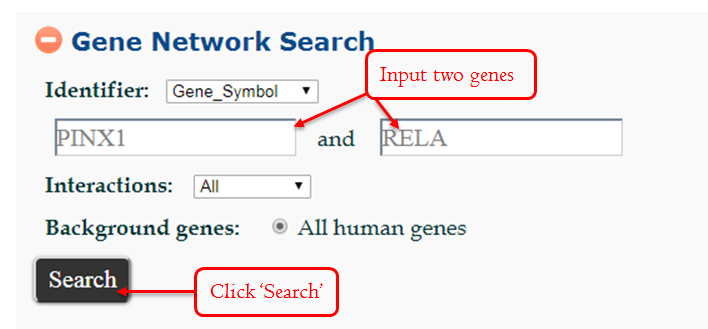
Function Related Gene and Gene Network
Basic rules
- Boolean operators: AND, OR, NOT. Boolean operators must be entered in uppercase letters. Space is treated as AND operator. Boolean logic refers to the logical relationships among search terms. Boolean operators are processed from left to right. Use parentheses to nest terms together so they will be processed as a unit.
- Search field tags: [S], [A], [G], [GS], [GA] . [S] and [A] are used to search any terms in the literature, [S] means search terms in the sentence, [A] means search terms in the abstract. [G], [GS] and [GA] means the term will be considered as a gene symbol, the synonyms of this gene will be searched. [G]/[GS] means search gene in sentence, [GA] means search gene in the abstract. The search field tag must follow the term. At most, you can use four tags. Multiple free terms that are wish to search in the same field are recommended to bring into one search string and then tag with the search field. The absence of a search tag will be considered as searching free text in the sentence.
- When input one gene symbol and tag [G] or [GS], the interactions of this gene will be shown in the result page.
- When you select to search in GeneRIF sentence, [A] and [GA] do not work, and ignored.
- Enclosing the phrase in double-quotes, such as “cancer stem cell”.
- Escaped characters: [ ] ( ) ” “, should be used in pairs.
- The length of a search term is limited in 3~500 characters.
- The result for too general terms may not be shown, such as cell, protein, gene, and so on.
Query Syntax
1. Single term search
- autophagy
- autophagy [S]
- Search for genes that co-occur with autophagy in the sentence. - autophagy [A] - Search for genes that co-occur with autophagy in abstract while search in MEDLINE.
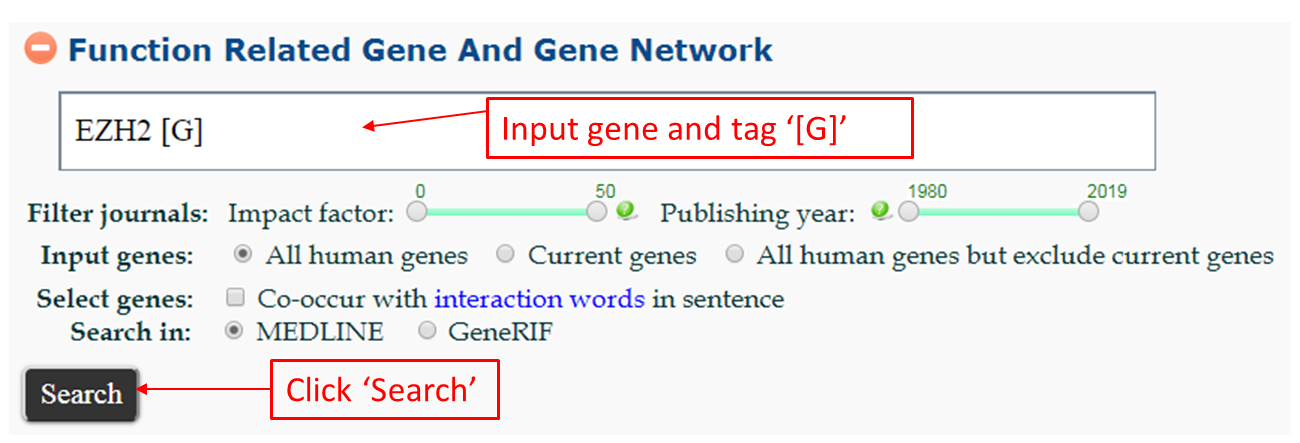
- EZH2 [G]
- EZH2 [GS]\ - Search for genes that co-occur with EZH2 and its synonyms in sentence. Also, this search will return high confidence interactors of EZH2.
- EZH2 [GA] - Search for genes that co-occur with EZH2 and its synonyms in MEDLINE abstract.
2. Phrase search
- “nasopharyngeal carcinoma” - Search for genes that co-occur with the phrase nasopharyngeal carcinoma in the sentence.
3. Boolean search
- “nasopharyngeal carcinoma” metastasis
-
“nasopharyngeal carcinoma” AND metastasis
- Two queries produce the same results which are genes that co-occur with both nasopharyngeal carcinoma AND metastasis in the sentence. -
“nasopharyngeal carcinoma” OR NPC - Search for genes that co-occur with nasopharyngeal carcinoma or NPC in the sentence.
-
NPC NOT “Niemann Pick type C” - Search for genes that co-occur with NPC but not with Niemann Pick type C in the sentence.
4. Query grouped terms
- (“nasopharyngeal carcinoma” OR NPC ) AND (“metastasis” OR “invasion”) * - Search for genes that co-occur with *nasopharyngeal carcinoma or NPC, and metastasis or invasion in the sentence.
5. Query tagged and grouped terms
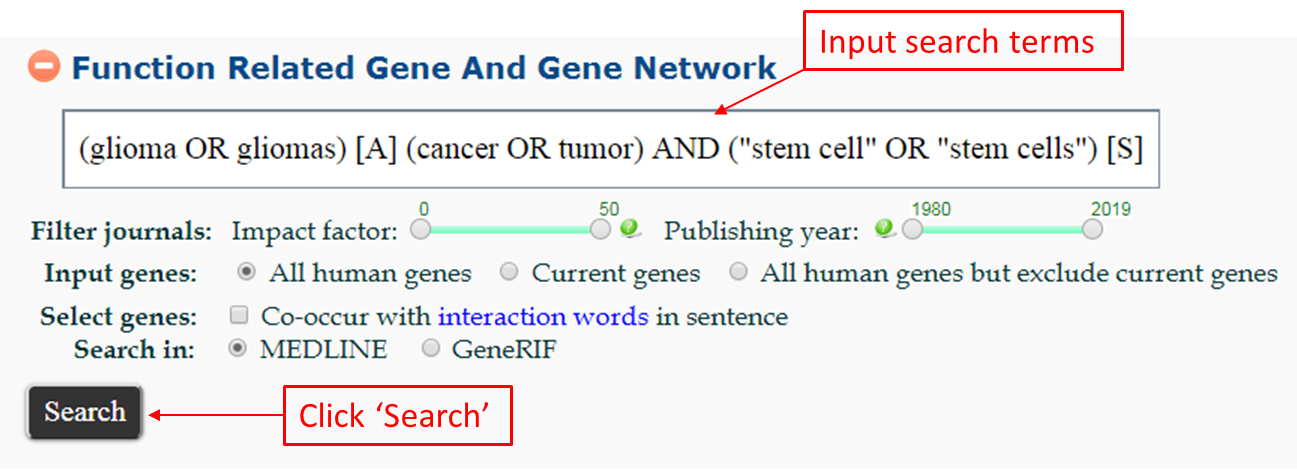
-
(glioma OR gliomas) [A] (cancer OR tumor) AND (“stem cell” OR “stem cells”) [S] - Search for genes that co-occur with glioma or gliomasin MEDLINE abstract, and stem cell or stem cells in MEDLINE sentence. This case is the default example that is shown in the input box, just click the search button.
-
target [S] MIR27A [G] - Search for genes that co-occur with target and MIR27A and its synonyms in sentence. While query target MIR27A, it will not consider the synonyms of MIR27A.
Tips of this module:
- Use “Filter journals”, extract genes and abstracts in high quality and the newest papers.
- While searching in the sentence, select “Co-occur with interaction words in sentence”, the relationship between gene and terms may be stronger.
- Use “Input genes”, to find related genes in your gene list, or to exclude related genes that you have known.
- Related genes that can not be found in the literature, can be added into the gene network if they have connections with other selected genes.
Details
Search terms related genes
Search gene-gene association
Result gene and literature page – term-gene associations

Result gene and literature page – gene-gene associations

Download genes/PMIDs or create gene network
GO / Pathway Analysis
GenCLiP 3 also integrates some canonical database to annotate genes. GO Analysis annotates terms of biological process and molecular function. Pathway Analysis annotates biomedical pathways from KEGG, REACTOME, and BIOCARTA, and so on, which were collected by ConsensusPathDB. The function and usage are similar to the keyword’s module, except editing. The two modules can help users to discover enriched GO terms and pathways.
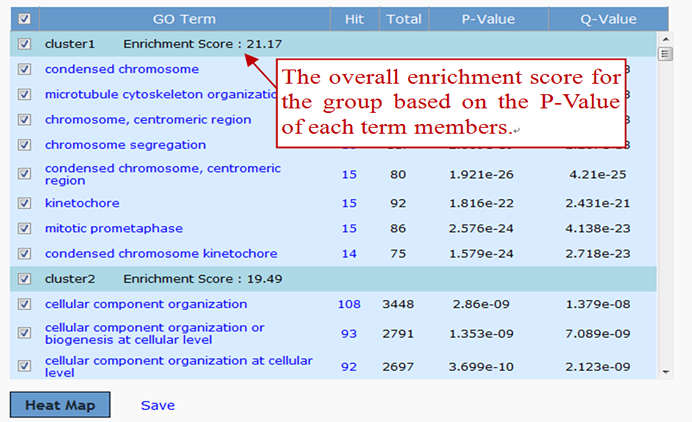
The group enrichment score, the geometric mean (in -log scale) of terms’ p-values in the cluster, is used to rank their biological significance. Thus, top-ranked annotation groups most likely have consistently lower p-values for their annotation terms.
Fuzzy Cluster
GenCLiP 3 provides three modules to identify the most relevant (over-represented) biological terms associated with a given gene list. Here, biological terms(annotation) include keywords derived from literature, GO terms(biological process and molecular function) and pathways(three resources collected by GSEA, including KEGG, BioCarta, RECTOME).
Generally, annotated biological terms are abundant and multifarious, bring an impending need for classifying them. Condensing large annotation lists into biologically meaningful modules greatly improves our ability to assimilate large amounts of information and thus switches functional annotation analysis from a gene-centric analysis to biological module-centric analysis. To identify the related biological terms can help biologists to assemble a bigger biological picture for a better understanding of biological themes.
DAVID proposed a novel clustering algorithm, named fuzzy cluster, to address this problem efficiently. Fuzzy cluster algorithm classifies highly related terms into functionally related groups. Typically, a biological term is a cooperation of a set of genes. As an example, if two or more biological processes are done by a similar set of genes, the processes might be related to the biological network somehow.
This algorithm adopts kappa statistics to quantitatively measure the degree of the agreement on how terms share similar participating genes for. After scanning all pairs of the given term to other terms, the closely related terms to the given one could be listed and sorted. Kappa result ranges from 0 to 1. The higher the value of Kappa, the stronger the agreement. Kappa more than 0.7 typically indicates that the agreement of two terms is strong. Kappa values greater than 0.9 are considered excellent. The higher the setting, the fewer terms will be put into a clustered group, which leads to a higher quality of functional classification results with fewer groups and fewer term members. Kappa value 0.3 starts giving meaningful biology based on DAVID’s genome-wide distribution study. Anything below 0.3 has a great chance to be noise.
Here we modify some steps of DAVID’s fuzzy cluster to algorithm adapt our approach. GenCLiP 3 uses a similar fuzzy clustering concept as functional classification by measuring relationships among the annotation terms based on the degree of their co-association with genes within the user’s list to cluster somewhat heterogeneous, yet highly similar annotation into functional annotation groups. The main steps as follows:
Step 1: The statistical significance of a term is assessed by the hypergeometric test, P<0.05 is considered significant. Measure the relationships of all term-term pairs with Kappa statistics. The threshold of kappa value is 0.45.
Step 2: Each term annotates several genes, array the numbers in ascending. From small to big, each term connects with their most closely related term which has the biggest Kappa value. And then, two single terms will be clustered in a group, or a single term will be put into a formed group. If two terms have been assigned into groups, they would not be merged.
Step 3: After cluster processing, the terms will be divided into groups or singles. Calculate the enrichment score of the group, which is the geometric mean of minus log transformation of the p-value of those terms involved in groups. Single terms are also scored by the minus log transformation of the p-value. The result table shows the groups and single terms by arraying the score in descending.
Key Points:
- Number of total groups is dynamically determined based on the given genes and different threshold (p-value, hit genes and total genes).
- Fuzziness: measure the relationship by shared identical genes.
- An annotation just belongs to one cluster or be a single.
This type of grouping of functional annotation can give a more insightful view of the relationships between annotation categories and terms compared with the traditional linear list of enriched terms, as highly related annotation terms may be dispersed among hundreds, if not thousands, of other terms. This reduces the burden of associating different terms associated with the similar biological process, thus allowing the biological interpretation to be more focused at a biological module level. Empty report means that there are no annotations passed the specified threshold. It does not mean that no annotation exists.
Login / Register
Click the Login text link at the top of the page opens a new page requesting your Email (username) and password. If you have not been previously registered with GenCLiP 3, there is a link to the register page. If you have already registered but forgotten your password, you can regain your password.
We try to make it as easy as possible for users to use GenCLiP 3. Therefore, registration is not forcibly required. Unregistered users have access to the same functionality as registered users. By registering, the user can retrieve and continue to analyze the recent analyses through click the job name on the right-hand side of the web page. For registered users, If your archive of analysis is inactive for more than one month, the system will clean up all information (your gene list, results and so on) on the server-side to conserve disk space.
